Preparation
The audience for this workshop session are bioinformaticians or life scientists interested in learning to use semi-automated workflow includes differential gene expression analysis, statistical analysis of metabolomics data, as well as pathway enrichment analysis for both transcriptomics and metabolomics data followed by integration of this data through network analysis to identify disease-related processes and visualization of multi-omics data . Participants should have some prior experience with R data analysis.
Follow the instructions to download install all required software tools for the workflow.
The workflow was created with the following versions of the software:
- R 4.1.3
- R-studio 2021.09.02
- Cytoscape v3.9.1
You can get the latest versions of each tool by the following links.
Step 1: Download required software tools
Step 2: Install required software tools
Step 3: Install SPARQL R package from source
For Windows user, due to ‘SPARQL’ was removed from the CRAN repository it should be install from a source
- Download the latest verison of the package
- Unzip “SPARQL_X.XX.tar.gz” to a location (path_to_file) where you can find it for the next step
- Run the following command in RStudio
install.packages(path_to_file, repos = NULL, type=”source”) —–Step 4: Install required apps in Cytoscape
- Open Cytoscape
- Go to the app manager: Apps -> App Manager
- Install the WikiPathways, stringApp and clusterMaker2 apps fiven in following figures
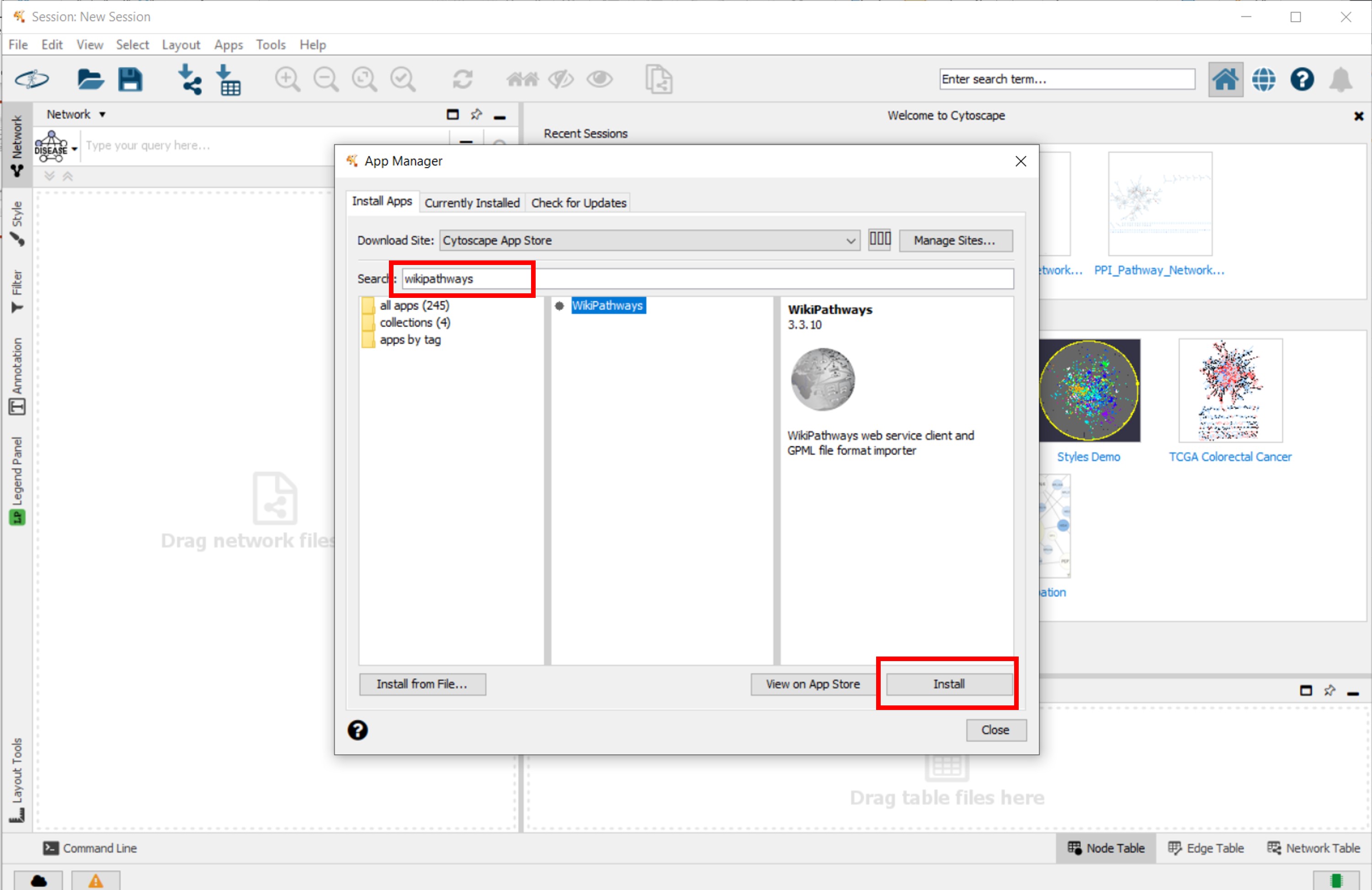 Figure 1 |
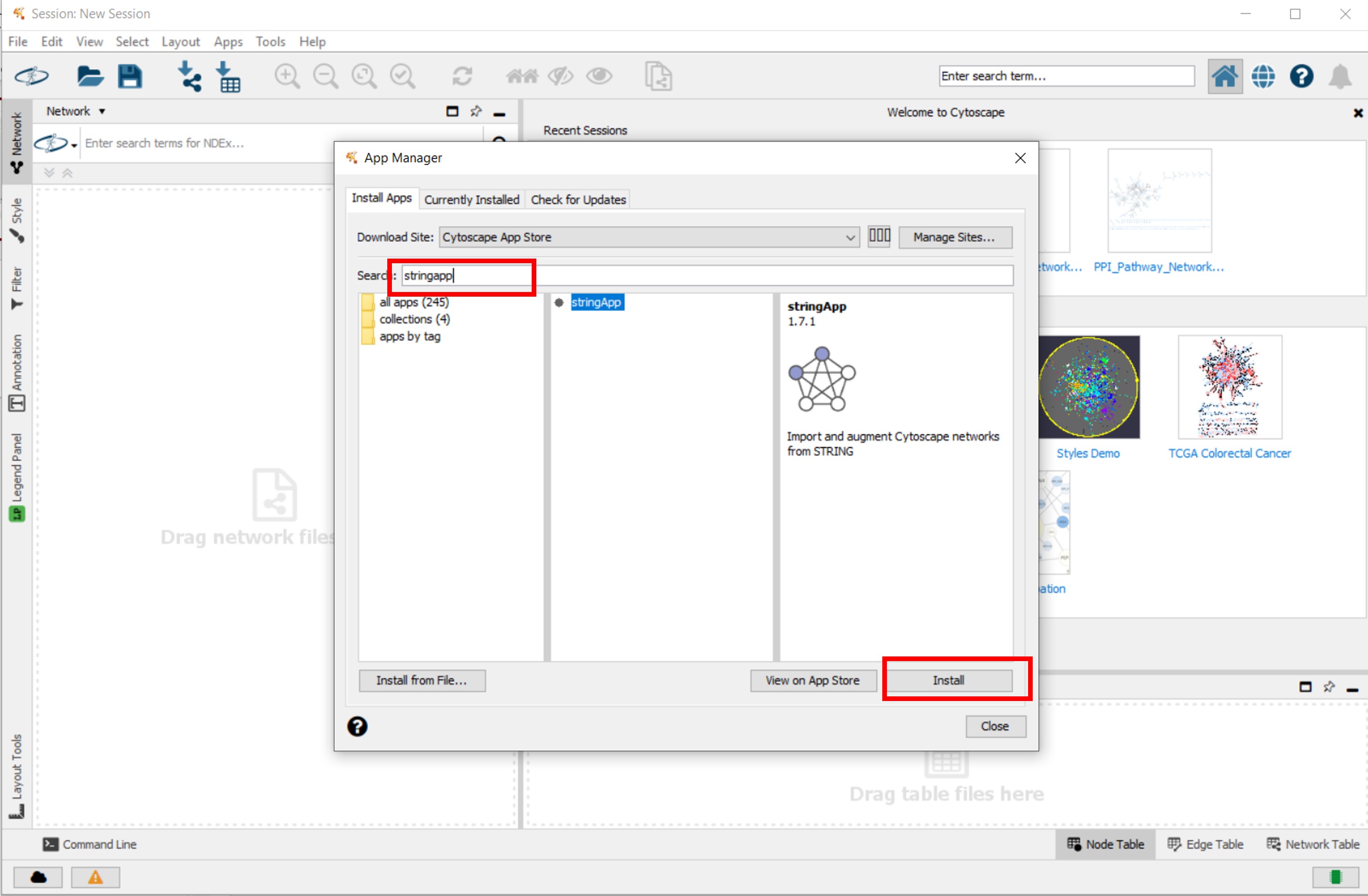 Figure 2 |
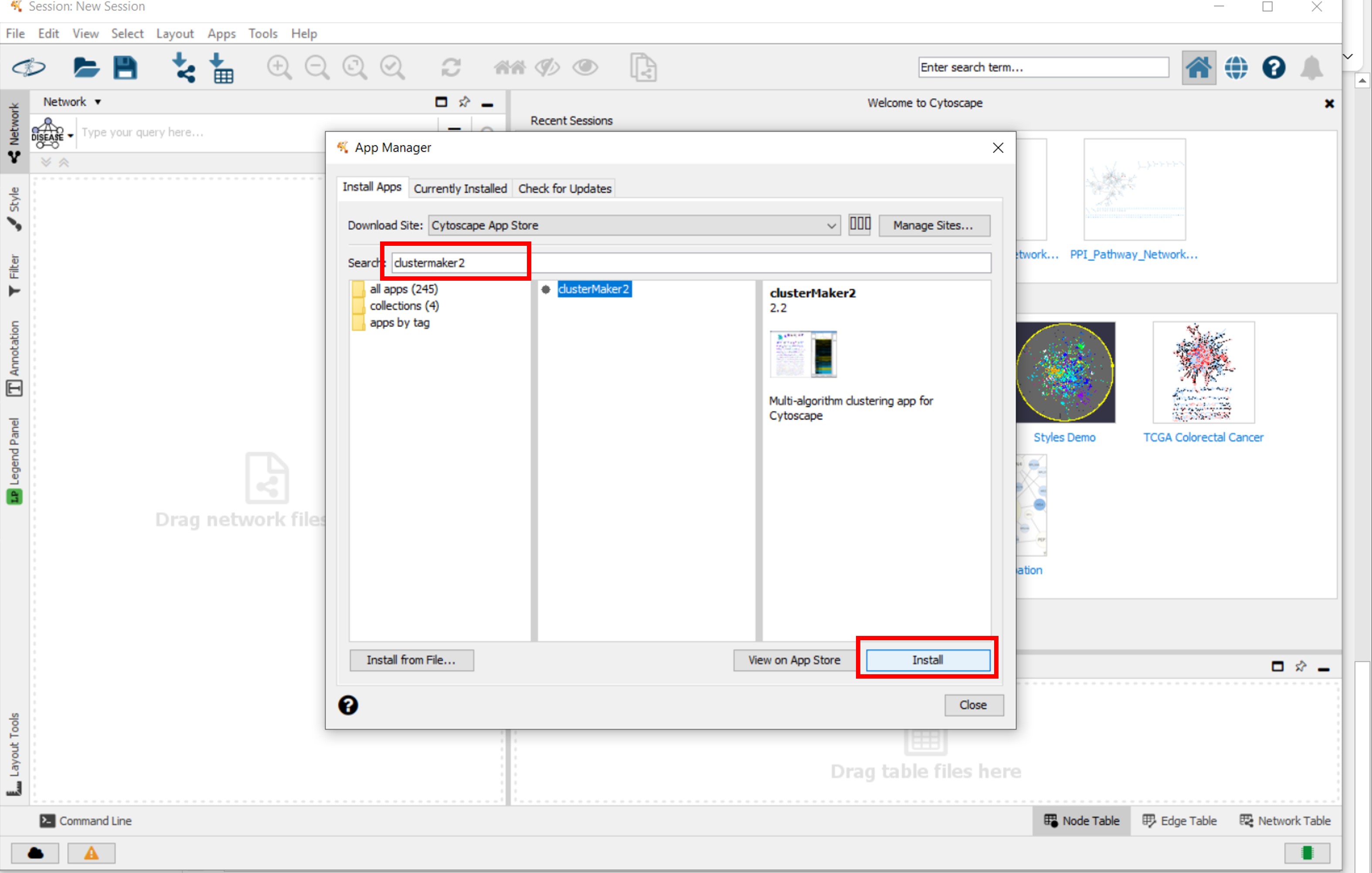 Figure 3 |